SARS-CoV-2 Variants
Our work with SARS-CoV-2 covers surveillance for new variants, understanding the neutralizing antibody responses in vaccinated and infected individuals as well as the basic virology and molecular biology of virus infection. Similar to our influenza surveillance efforts, we focus on the isolation of SARS CoV-2 from the clinical specimens using our biosafety level 3 (BSL-3) high containment facility so we can both generate virus isolates for further characterization as well as to measure the infectious virus load.
The SARS-CoV-2 isolates are then characterized for their replication in standard cell cultures like Vero-TMPRSS2 cells or in our primary hNEC culture system. We have focused particularly on the replication of SARS-CoV-2 variants at physiologically relevant temperature ranges (32°C-37°C) and have shown that variants display different temperature sensitivities.
We are also interested in SARS-CoV-2 Spike protein functions (receptor binding, proteolytic cleavage and fusogenic activity) and how those functions contribute to virus fitness. We have also identified variants that have Spike protein mutations that are also found in variants of concern, but whose case numbers have never risen to levels that would make them significant risks to humans.
Understanding what viral genetic factors are missing from these variants that contributed to their reduced spread will help us to determine how mutations outside of the Spike protein may contribute to virus fitness and spread in the human population.
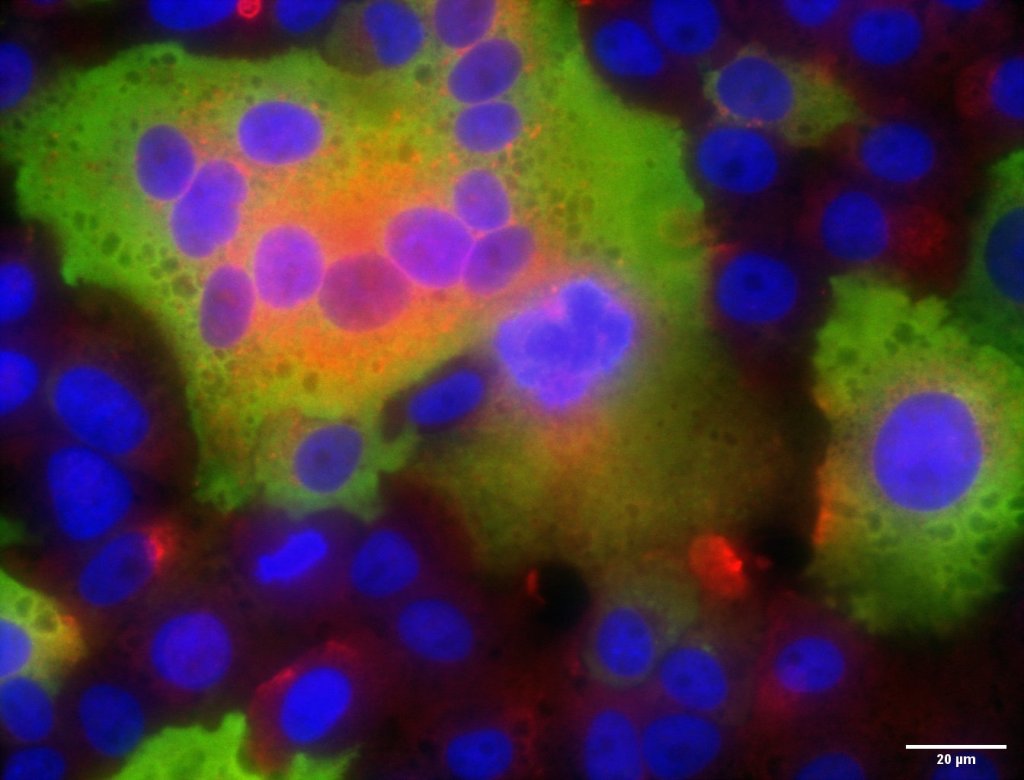
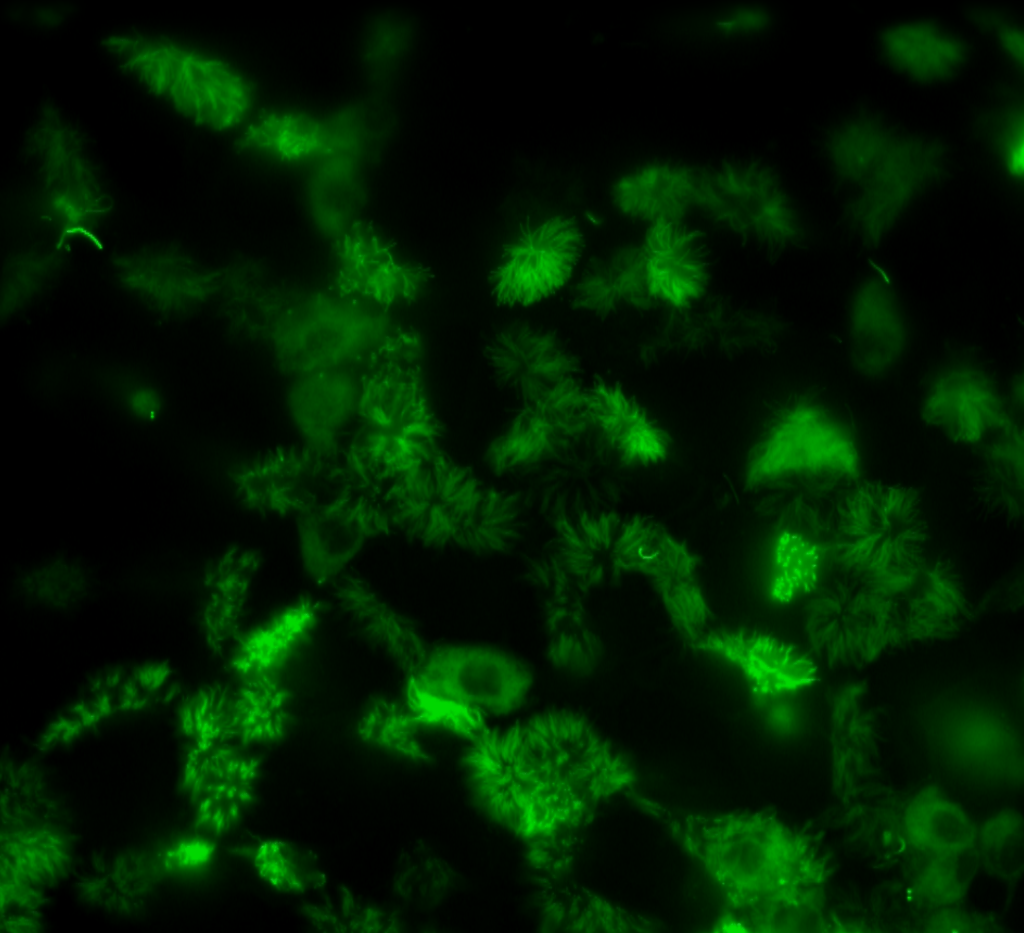
Syncitial cells after infection with the Delta variant, nuclei stained in blue, Nucleoprotein stained in green and staining using purified IgG from convalescent plasma in red. Source: Dr. Eddy Anaya 
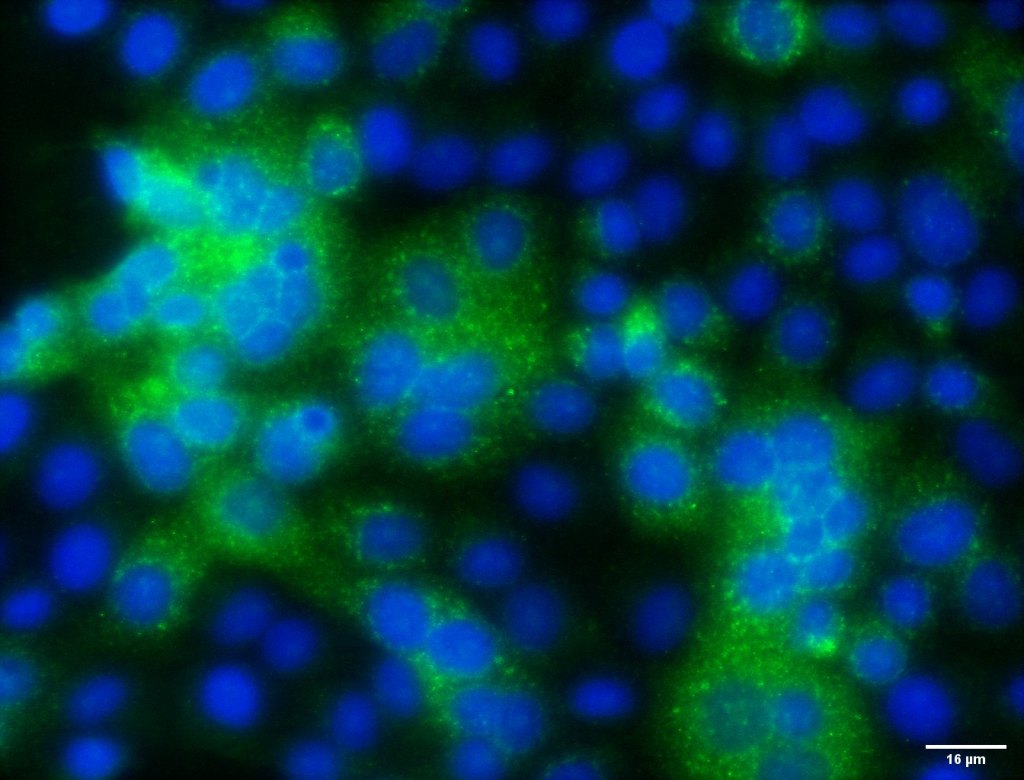
Syncytial cells after infection with the Delta variant, nuclei stained blue and spike protein stained green. Source: Dr. Eddy Anaya
Human Nasal Epithelial Cell (hNEC) Investigations
The human respiratory epithelium is the primary portal of entry for respiratory viruses and serves as the first line of defense against several invading respiratory viral pathogens. In addition to the virus characterization research we conduct, the lab is also interested in host-cellular responses to viruses. There are multiple ongoing projects that investigate the host cell responses to viral infection which use a combination of in vitro mammalian and ex vivo primary human nasal/bronchial epithelial cell cultures.
Mitochondrial dynamics in the context of temperature and viral infection.
We have previously shown that respiratory tract temperature has been shown to influence host immune responses, viral replication rates, and viral titers during influenza A virus infection. However, the underlying mechanisms mediating these differences and the effect of respiratory tract temperature on epithelial cell function are not well understood. We use a variety of microscopy-based assays, metabolic assays, and viral assays to determine how components of host cell biology, like the mitochondrial compartment, influence the ability of epithelial cells to support virus replication and initiate effective immune responses across the respiratory tract.
Transport of IgG/IgA across respiratory epithelial cell barriers
The neutralizing activity in human serum is considered the best correlate of protection from SARS-CoV-2 infection and disease. However, it remains unclear how and which specific antibody subtypes present in serum can be effectively secreted into the lumen of the upper and lower respiratory tract, where antibodies are needed to protect from infection.
Using human respiratory epithelial cell cultures derived from different parts of the respiratory tract, we were able to determine differences in IgG and IgA transcytosis in the upper and lower respiratory tract and their effectiveness at neutralizing SARS-CoV-2 and Influenza.
Live Attenuated Influenza Vaccine
From 2013-2016, the H1N1 component of the live, attenuated influenza vaccine (LAIV) performed very poorly in contrast to the inactivated influenza vaccine, even though the two were antigenically matched. Using a primary, differentiated human nasal epithelial cell (hNEC) culture system, we have been able to assess fitness differences between isogenic LAIVs containing HA segments derived from either “successful” or “failed” vaccines that differ by as few as 4 amino acids.
These viruses show little differences in fitness when grown on MDCK cells, but large differences when grown on hNECs. We have found that better vaccine viruses show markedly improved replication in hNEC cultures at 32oC and 37oC, while failed vaccines are unable to replicate on hNECs at 37oC, showing that including temperature while evaluating candidate viruses gives us more power to detect differences in viral replication in ways that may correlate with vaccine efficacy.
We have also identified subtype specific mutations in the M2 protein cytoplasmic tail that influence the temperature sensitivity of these viruses. Future work in the lab is focusing on which specific HA mutations contribute to these differences in viral fitness as well as identifying specific mutations in the A/Ann Arbor LAIV backbone that can broadly improve LAIV fitness regardless of donor virus characteristics.

Filamentous influenza budding from a cell (source: Dr. Andrew Pekosz)
H3N2 Reassortment
Surveillance for influenza viruses is essential for choosing virus strains for use in influenza vaccine formulations which change almost yearly based on antigenic drift in circulating virus strains. The Johns Hopkins Center of Excellence in Influenza Research and Response (JH-CEIRR) has been doing influenza surveillance in Baltimore, MD, Taipei, Taiwan and Macha Zambia since 2013 with the goal of characterizing the antigenic structure and replication fitness of Influenza A and B Viruses and linking that virological data to clinical and demographic data from infected individuals.
The 2017-18 influenza season in the United States (US) was characterized by high amounts of influenza illness and mortality. Based on viral whole genome sequencing analysis of clinical samples, we identified two major reassortment events within Influenza A/H3N2 viruses in the 2017-18 season, resulting in viruses of the HA clade 3C.2a2 and clade 3C.3a with novel gene segment constellations, some segments of which were derived from HA clade 3C.2a1 viruses.
We used in vitro mammalian cell culture and ex vivo primary human nasal epithelial cell (hNEC) system to characterize viruses and found the 2017-18 dominant virus, reassortant 3C.2a2 viruses, replicated with faster kinetics and to a higher peak titer when compared to the parental 2016-17 3C.2a2 and 3C.2a1 viruses, suggesting reassortment led to an increased production of infectious virus particles. The results support demographic and clinical analysis that patients infected with the reassortant 3C.2a2 viruses had a higher severity score compared to patients infected with parental 3C.2a2.
The 2017-18 reassortant 3C.3a viruses did not show a replication advantage compared to the parental 3C.3a virus, but they were able to escape inhibition by anti-NA antibodies because the 3C.2a1 virus donated an NA segment that had a glycosylation site that masked a known NA neutralizing antibody epitope. Demographic analysis showed that the parental 3C.3a viruses were found to infect a large number of pediatric patients in 2016-17. However, the 3C.3a reassortant viruses was able to infect populations over 18 years of age in 2017-18 and became the dominant H3N2 in the US and Europe the following year.
Genomics Core
Additional information coming soon 🙂
